FDA will not issue registration certificates to medical device companies, and will not issue confirmation certificates for products or companies that have been registered or listed. Enterprise registration and product listing information does not mean that FDA approved the company and its products.
FDA registration issues that we generally need to pay attention to are as follows:
Question 1: Which agency issued the FDA certification?
Answer: There is no certification for FDA registration. After product pass registered the with FDA, it will get the registration number, and FDA will give the applicant a reply.But there is no such thing as an FDA certification. FDA issued such a notice at this time is a strong reminder! Due to the recent development of the epidemic in the United States, the demand for medical anti-epidemic products exported to the United States has increased significantly, and the demand for export registration has also increased.However, some companies pretend to be FDA and issue certification to manufacturers, and when some distribution companies consult manufacturers. they may get the fake "FDA certificates" .
Question 2: Does FDA need to be tested by a designated certification laboratory?
Answer: FDA is an enforcement agency, not a service agency.If someone says they are belong to FDA certified laboratories, then he is at least misleading consumers. Because FDA has neither a service-oriented certification organization and laboratory for the public, nor a so-called "designated laboratory".As a federal law enforcement agency, the FDA cannot engage in such a matter of being a referee and an athlete at the same time.FDA will only recognize the GMP quality of service testing laboratories and issue qualified certification.But it will not "designate" the public or recommend a specific one company or other companies.
Question 3: Does the FDA registration require a US agent?
Answer: Yes, companies must designate an American citizen(company / society) as their agent when registering with the FDA. The agent is responsible for the process services in the United States and is the medium for contacting FDA and the applicant.
Common misunderstandings of FDA registration
1.FDA registration and CE certification are different,its certification mode is different from the CE certification product testing + report certificate mode,FDA registration actually uses the integrity declaration model. That is: you are responsible for your products complying with relevant standards and safety requirements.If something goes wrong with the product, then it has to bear corresponding responsibilities.
2.FDA registration validity period:FDA registration is valid for one year, if more than one year, you need to resubmit the registration, and pay for the annual fee.
3.Is there a certificate for FDA registration? In fact, there is no certificate for FDA registration.

According to the different risk levels, the FDA 510(k) certification for medical devices in the United States divides medical devices into three categories(Ⅰ, II, and III), with the highest risk category III.
FDA clearly stipulates its product classification and management requirements for each medical device. At present, there are more than 1,700 kinds of FDA medical device product catalogs.Any medical device that wants to enter the US market must first clarify the classification and management requirements for the product to be listed.For any product, the enterprise needs to carry out enterprise registration and product listing.
For Class I products(accounting for about 47%), general control is implemented.Most products only need to register, list and implement GMP regulations.Products can enter the US market(of which a few products even GMP Also exempt.Very few reserved products need to submit a 510(K) application to the FDA, namely PMN.(Premarket Notification)
For category II products(accounting for about 46%), special control is implemented.After the registration and listing, the enterprise needs to implement GMP and submit 510(K) application(very few products are 510(K) exempt)
For Class III products(approximately 7%), pre-market licensing is implemented.After registration and listing, enterprises must implement GMP and submit PMA(Premarket Application) application to FDA(Part III)Products are still PMN).
For class Ⅰ products, after the company submits relevant information to FDA, FDA only makes announcements, and no relevant documents are issued to the company.FDA will give the company a formal market access approval letter at the same time as the announcement.That is, companies are allowed to sell their products directly on the US medical device market in their own name.
As for whether to go to the enterprise for on-site GMP assessment during the application process, it is up to the FDA to decide on comprehensive factors such as product risk level, management requirements and market feedback.
Based on the above, it can be seen that most products can obtain FDA certification after enterprise registration, product listing, and implementation of GMP for medical devices, or after submitting a 510(K) application.
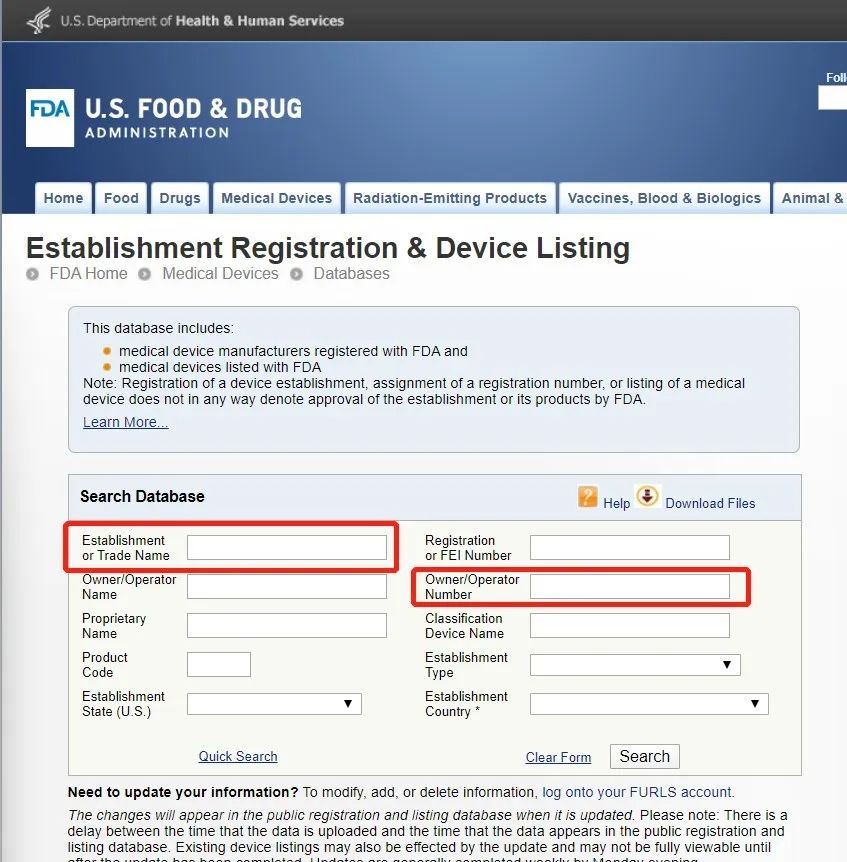
How to check if the product has obtained FDA listing or 510K registration?
The only authoritative way: check on the FDA official website
Last: The Ministry of Commerce Will Work with Relevant Departments to Strengthen the Quality Management of Medical Next: The Last
Copyright @ 2016 NANCHANG MICARE MEDICAL EQUIPMENT CO.,LTD. E-mail:info@micare.cn
Add:Yaohu West 5th Road,Hi-Tech Zone, Nanchang,Jiangxi,China